
Ein experimenteller, dreiteiliger Ansatz bei Lungenfibrose: direkt, regenerativ und krankheitsmodifizierend
Lungenfibrose bleibt trotz moderner antifibrotischer Medikamente eine Erkrankung mit hohem ungedecktem medizinischem Bedarf.
Die zugelassenen Standardtherapien Nintedanib und Pirfenidon können den Verlust der Lungenfunktion verlangsamen, aber Fibrose in der Regel nicht stoppen oder strukturell rückgängig machen.
Im ANOVA Institute for Regenerative Medicine in Offenbach/Main wird jetzt ein neuartiger, experimenteller, kombinierter Therapieansatz verfolgt. ANOVA verfügt als Deutschlands erste Institution über eine Herstellungsgenehmigung für mesenchymales Stammzellsekretom und damit über eine besondere Ausgangsposition: Ein zellfreier, biologisch aktiver Regenerationsansatz kann mit etablierten antifibrotischen und senolytischen Mechanismen kombiniert werden. Das angestrebte Ziel ist nicht nur, Fibroseprogression zu bremsen, sondern das fibrotische Milieu der Lunge biologisch umzuprogrammieren.
Der vorgeschlagene Ansatz kombiniert drei komplementäre Wirkprinzipien:
1. Antifibrotikum als Krankheitsbremse
Nintedanib, Pirfenidon — und perspektivisch Nerandomilast — adressieren zentrale pro-fibrotische Signalwege und reduzieren den jährlichen FVC-Verlust. Nerandomilast, ein selektiver PDE4B-Inhibitor, wurde in den USA 2025 für IPF zugelassen; laut FDA war dies die erste neue IPF-Therapie seit mehr als zehn Jahren.
2. Mesenchymales Stammzellsekretom als experimenteller lokaler Regenerationsimpuls.
Das inhalative Aerosol bringt potentiell bioaktive Faktoren, extrazelluläre Vesikel, miRNAs und immunmodulierende Signale direkt an den Ort der Pathologie: Alveolarepithel, interstitielle Fibroblasten, Makrophagen und die gestörte epithelial-mesenchymale Nische. Erste externe experimentelle klinische Daten aus kleinen Studien mit nebulisierten hUCMSC-EVs bei pulmonaler Fibrose deuten auf Sicherheit und mögliche Wirksamkeit hin, bleiben aber explorativ.
3. Senolytika als Reset des fibrotischen Mikromilieus.
Seneszente Zellen gelten als Treiber chronischer Entzündung, SASP-Signalgebung, Fibroblastenaktivierung und gestörter Gewebereparatur. Die Kombination Dasatinib + Quercetin wurde in IPF bereits in einer Phase-I-Pilotstudie untersucht; sie war machbar und lieferte frühe Signale, ist aber nicht als IPF-Therapie zugelassen.
Die Hypothese: Das Antifibrotikum bremst das Fortschreiten der Lungenfibrose, Senolytika entfernen pathologische Signaltreiber und das mesenchymale Stammzellsekretom unterstützt Reparatur und Immunmodulation. Gerade die inhalative Applikation des Sekretoms ist strategisch attraktiv: hohe lokale Exposition, direkter Zugang zum Lungengewebe, kleines Kompartiment mit hoher Dosis und Vermeidung des Abbaus im Blut.
Der bevorzugte Entwicklungsweg wäre ein add-on Design auf zugelassener Basistherapie, etwa Nintedanib oder Pirfenidon, mit einem sequenziellen oder zyklischen Senolytikum-Regime und begleitender inhalativer Sekretomgabe.
Um mögliche Nebenwirkungen frühzeitig zu erkennen und zu behandeln, würde die Behandlung unter engmaschiger Überwachung während eines kurzen stationären Aufenthaltes erfolgen. Dabei würden Lebertoxizität, Blutungsrisiko, Pleuraergüsse, pulmonale Hypertonie, Infektanfälligkeit und pharmakokinetische Interaktionen überwacht; Dasatinib ist ein onkologischer Tyrosinkinaseinhibitor mit relevanten Risiken, während Nintedanib ebenfalls Leber- und Blutungsrisiken aufweist.
Die klinische Vision ist ein multimodaler, lokal verstärkter, biologisch rationaler Kombinationsansatz für Patienten mit IPF oder progressiver pulmonaler Fibrose: Standard-of-care plus gezielte Seneszenz-Elimination plus inhalatives regeneratives Sekretom — mit dem Anspruch, von reiner Progressionsverlangsamung in Richtung Krankheitsmodifikation zu gehen.
Im ANOVA Institute for Regenerative Medicine setzen wir seit vielen Jahren auf den Fortschritt mit Hilfe der experimentellen, translationalen Medizin. Wir gehören zu den Pionieren der Stammzelltherapie in Europa und verfolgen vielfältige Ansätze der regenerativen Medizin. Für uns sind innovative Therapieansätze keine Bedrohung – sondern eine faszinierende Welt neuer Möglichkeiten für unsere Patienten.
Wenn Sie an Lungenfibrose leiden und mehr über experimentelle, individuell abgestimmte Therapieansätze erfahren möchten, sprechen Sie mit uns. Nach einer individuellen ärztlichen Beurteilung besprechen wir, ob ein solcher Ansatz für Ihre Situation infrage kommt.
Wissenschaftliche Informationen
Lungenfibrose ist selten, aber verheerend. Allein die idiopathische pulmonale Fibrose betrifft Schätzungen zufolge rund 3 Millionen Menschen weltweit; in den USA leben mehr als 250.000 Menschen mit pulmonaler Fibrose oder interstitieller Lungenerkrankung. Für Deutschland zeigen Kassendaten zur IPF eine Punktprävalenz von etwa 24 pro 100.000 Einwohner, was grob rund 20.000 IPF-Patienten entspricht — hinzu kommen weitere Formen progressiver Lungenfibrose.
1. Medizinischer Hintergrund und therapeutische Lücke
Lungenfibrose ist selten, aber verheerend. Allein die idiopathische pulmonale Fibrose betrifft Schätzungen zufolge rund 3 Millionen Menschen weltweit; in den USA leben mehr als 250.000 Menschen mit pulmonaler Fibrose oder interstitieller Lungenerkrankung. Für Deutschland zeigen Kassendaten zur IPF eine Punktprävalenz von etwa 24 pro 100.000 Einwohner, was grob rund 20.000 IPF-Patienten entspricht — hinzu kommen weitere Formen progressiver Lungenfibrose.
Die Erkrankung ist inzwischen auch einer breiteren Öffentlichkeit bekannt, seit bei Kronprinzessin Mette-Marit von Norwegen 2018 eine chronische Lungenfibrose diagnostiziert wurde.
Für Patienten im Endstadium bleibt die Lungentransplantation oft die einzige lebensverlängernde Option. Interstitielle Lungenerkrankungen einschließlich Lungenfibrose gehören weltweit zu den wichtigsten Indikationen für Lungentransplantationen; dennoch kommt nur ein kleiner Teil der Patienten dafür infrage, weil Alter, Begleiterkrankungen, Organmangel und Krankheitsdynamik die Auswahl stark begrenzen. Eine deutsche Analyse berichtete, dass IPF nach Einführung des Lung Allocation Score einen deutlich größeren Anteil der transplantierten Hochdringlichkeitsgruppe ausmachte — etwa 27 % in der Kategorie D.
Die Transplantation kann lebensrettend sein, ist aber hochriskant: Es handelt sich um einen großen Eingriff mit lebenslanger Immunsuppression, Infektionsrisiko, Abstoßungsreaktionen, chronischer Transplantatdysfunktion und begrenzter Langzeitprognose. Neuere Übersichten nennen für Lungentransplantierte insgesamt eine mediane Überlebenszeit von etwa 6,2 Jahren, für interstitielle Lungenerkrankungen etwa 5,2 Jahre. Genau deshalb besteht ein dringender Bedarf an Therapien, die früher ansetzen, Progression bremsen, Gewebe schützen und eine Transplantation möglichst hinauszögern oder vermeiden helfen.
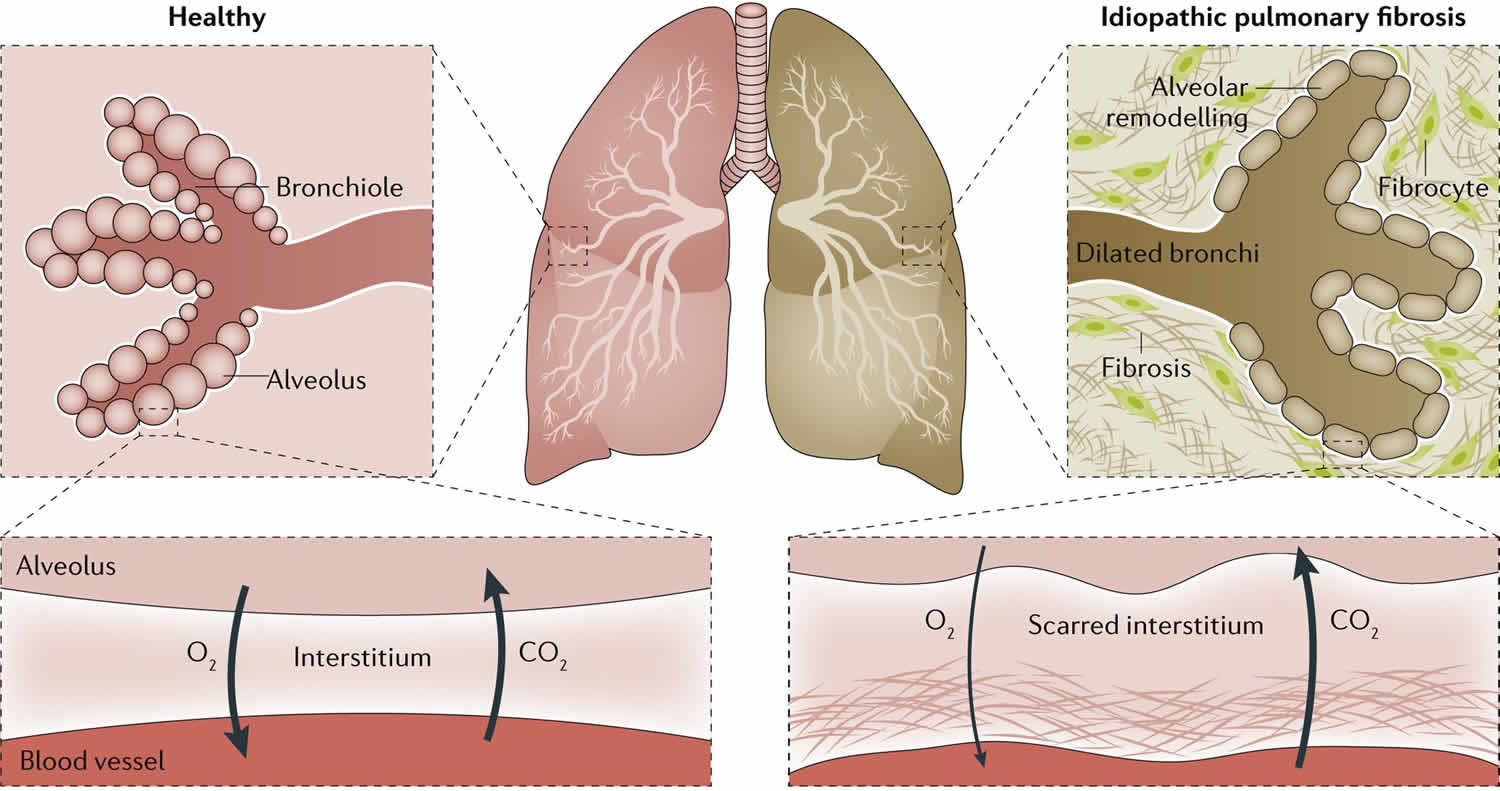
Idiopathische pulmonale Fibrose und andere progressive fibrosierende interstitielle Lungenerkrankungen sind durch eine fehlgeleitete Reparaturreaktion des Lungengewebes gekennzeichnet. Wiederholte mikroskopische Schädigungen des Alveolarepithels führen zu epithelialer Dysfunktion, Aktivierung von Fibroblasten und Myofibroblasten, überschießender extrazellulärer Matrixablagerung, Architekturverlust und zunehmendem Gasaustauschdefizit. Klinisch resultieren progrediente Dyspnoe, Husten, Belastungsintoleranz, Exazerbationen und reduzierte Überlebenszeit.
Die aktuellen Leitlinien für IPF und progressive pulmonale Fibrose stützen sich auf antifibrotische Therapien. Die ATS/ERS/JRS/ALAT-Leitlinie von 2022 behandelt IPF und PPF gemeinsam und gibt für Nintedanib bei PPF eine konditionale Empfehlung; für Pirfenidon bei PPF wurde weiterer Forschungsbedarf formuliert. In Europa ist Ofev/Nintedanib zugelassen für Erwachsene mit IPF sowie für andere chronische fibrosierende ILDs mit progressivem Phänotyp; Esbriet/Pirfenidon ist zugelassen für erwachsene Patienten mit IPF.
Trotz dieser Fortschritte bleibt die therapeutische Wirkung begrenzt: Antifibrotika verlangsamen den FVC-Verlust, führen aber typischerweise nicht zu einer robusten Regeneration zerstörter alveolärer Strukturen. Ein moderner Entwicklungsansatz sollte deshalb nicht nur einen einzelnen pro-fibrotischen Signalweg hemmen, sondern mehrere pathobiologische Ebenen adressieren: Fibroblastenaktivierung, seneszente Zellpopulationen, inflammatorische Fehlsteuerung, epitheliale Reparatur und lokale Gewebemikroumgebung.
2. Rationale für eine Kombination aus Antifibrotikum, Sekretom und Senolytikum
Die vorgeschlagene Kombination ist biologisch plausibel, weil die drei Komponenten unterschiedliche, aber komplementäre Ebenen der Fibrose adressieren.
2.1 Antifibrotische Basistherapie
Nintedanib hemmt mehrere Tyrosinkinasen, darunter Signalachsen über VEGF-, FGF- und PDGF-Rezeptoren, die an Fibroblastenproliferation, Migration, Myofibroblastendifferenzierung und Matrixproduktion beteiligt sind. Es ist daher besonders geeignet als Basistherapie bei IPF und bei progressiver fibrosierender ILD. Die EMA-Produktinformation nennt als empfohlene Dosis 150 mg zweimal täglich, mit 100 mg zweimal täglich als Option bei Unverträglichkeit.
Pirfenidon wirkt antifibrotisch und antiinflammatorisch, unter anderem über Modulation von TGF-β-assoziierten Mechanismen, Kollagensynthese und oxidativem Stress. Es ist in Europa für erwachsene Patienten mit IPF zugelassen.
Nerandomilast ist die neueste relevante Entwicklung. Der selektive PDE4B-Inhibitor zeigte in Phase-III-Daten bei IPF eine geringere Abnahme der FVC gegenüber Placebo und wurde im Oktober 2025 von der FDA für erwachsene Patienten mit IPF zugelassen; die FDA-Zulassung nennt 18 mg zweimal täglich. Für ein deutsches/ europäisches Konzept muss der regulatorische Status jedoch sauber geprüft werden; die EMA-Orphan-Designation für Nerandomilast bei IPF wurde 2025 auf Antrag des Sponsors aus dem EU-Register zurückgezogen, was nicht gleichbedeutend mit einer EU-Zulassung ist.
Für ein kurzfristig realisierbares deutsches Entwicklungsprogramm wäre daher Nintedanib der robusteste Kombinationsanker, insbesondere bei progressiven fibrosierenden ILDs. Pirfenidon wäre eine Alternative bei IPF. Nerandomilast ist wissenschaftlich hochinteressant, aber für ein EU/Germany-Konzept aktuell eher als perspektivische Option oder internationale Referenz einzuordnen.
3. Mesenchymales Stammzellsekretom: zellfreie Regeneration und Immunmodulation
Mesenchymale Stromal-/Stammzellen wirken in vielen präklinischen Modellen weniger durch dauerhafte Zellintegration als durch parakrine Signale. Das therapeutisch relevante Prinzip liegt im Sekretom: lösliche Wachstumsfaktoren, Zytokine, Chemokine, Lipide, Proteine, mRNA/miRNA und extrazelluläre Vesikel. Ein zellfreies Sekretom kann theoretisch mehrere Vorteile gegenüber lebenden Zellen bieten: bessere Standardisierbarkeit, geringeres Risiko unkontrollierter Zellpersistenz, bessere Lagerbarkeit, dosierbare biologische Aktivität und bessere regulatorische Charakterisierung.
Für pulmonale Fibrose ist die inhalative Gabe besonders attraktiv. Während systemische Zell- oder Vesikeltherapien von Biodistribution, pulmonalem First-Pass-Effekt und immunologischer Clearance abhängen, erlaubt ein Aerosol die direkte Deposition im Zielorgan. Pathophysiologisch relevante Zielstrukturen sind das Alveolarepithel Typ I/II, aktivierte interstitielle Fibroblasten, alveoläre Makrophagen, Endothelzellen und die extrazelluläre Matrixnische. Die zentrale Hypothese lautet: Lokales Sekretom kann die entzündliche und pro-fibrotische Mikroumgebung modulieren, epithelialen Stress reduzieren, Makrophagenpolarisation beeinflussen und Reparaturprogramme fördern.
Eine 2025 publizierte klinische Untersuchung zu nebulisierten humanen Nabelschnur-MSC-EVs berichtete bei pulmonaler Fibrose frühe Hinweise auf Sicherheit und mögliche klinische Aktivität; zwei Patienten mit fortgeschrittener post-inflammatorischer Fibrose zeigten in seriellen CT-Untersuchungen eine klinisch relevante Regression. Diese Daten sind vielversprechend, aber explorativ und nicht ausreichend, um Wirksamkeit als gesichert anzusehen. Reviews zu MSC-EVs betonen weiterhin präklinische Unterstützung, aber auch wesentliche offene Fragen zu Dosierung, Potenzassays, Langzeitsicherheit, Herstellbarkeit und klinischer Translation.
Für ANOVA ist die Herstellungsgenehmigung ein strategischer Vorteil, weil die Translation solcher biologischen Produkte wesentlich von GMP-naher Prozesskontrolle abhängt: Zellquelle, Kulturbedingungen, Sekretom-Ernte, Aufreinigung, Konzentration, Sterilität, Endotoxin, Partikelcharakterisierung, Proteom/miRNA-Profil, Freigabekriterien und Bioaktivitätsassays müssen reproduzierbar definiert sein. Für ein inhalatives Produkt kommen zusätzlich Aerosolphysik, Partikelgrößenverteilung, Verneblerkompatibilität, Stabilität nach Nebulisierung und Deposition im distalen Lungenkompartiment hinzu.
4. Senolytika: Dasatinib + Quercetin als pathobiologischer Reset
Seneszente Zellen sind metabolisch aktiv, teilungsunfähig und sezernieren ein proinflammatorisches, matrixmodulierendes Sekretionsprofil, das als SASP — senescence-associated secretory phenotype — bezeichnet wird. In der Lungenfibrose können seneszente Epithelzellen, Fibroblasten und Immunzellen die chronische Gewebedysfunktion aufrechterhalten. Seneszente Zellen produzieren unter anderem inflammatorische Zytokine, Wachstumsfaktoren und Matrix-remodellierende Enzyme, die Fibroblastenaktivierung, Epithelstress und Immunfehlsteuerung verstärken können.
Die Kombination Dasatinib + Quercetin ist eines der bekanntesten experimentellen senolytischen Regime. Dasatinib ist ein zugelassener onkologischer Tyrosinkinaseinhibitor, unter anderem gegen BCR-ABL und Src-Familienkinasen; Quercetin ist ein Flavonoid mit Einfluss auf anti-apoptotische und metabolische Signalwege. In IPF wurde D+Q bereits in einer randomisierten Phase-I-Pilotstudie geprüft, die Machbarkeit und Verträglichkeit untersuchte; die Evidenz bleibt jedoch frühphasig und reicht nicht für eine zugelassene IPF-Indikation.
Die Kombination mit Sekretom ist konzeptionell interessant: Senolytika könnten pathologische, pro-fibrotische Signalquellen reduzieren, während das Sekretom anschließend ein reparatives, immunmodulierendes Umfeld unterstützt. Eine mögliche Sequenz wäre deshalb nicht zwingend simultan, sondern zyklisch oder sequenziell: Zunächst Stabilisierung mit Antifibrotikum, dann kurze Senolytika-Impulse zur Reduktion seneszenter Zelllast, flankiert oder gefolgt von inhalativem Sekretom zur Unterstützung regenerativer Programme.
5. Wahl des Antifibrotikums für die Kombination
Für ein translationales Programm in Deutschland erscheint Nintedanib als bevorzugter Kombinationspartner, aus vier Gründen:
Erstens ist Nintedanib sowohl bei IPF als auch bei anderen chronischen fibrosierenden ILDs mit progressivem Phänotyp in Europa etabliert. Zweitens adressiert es Wachstumsfaktor-getriebene Fibroblastenaktivierung und ergänzt damit die immunmodulatorische und regenerative Logik des Sekretoms. Drittens ist die klinische Erfahrung bei progressiven fibrosierenden Erkrankungen breit. Viertens lässt sich ein add-on Studiendesign auf Nintedanib-Basis regulatorisch besser begründen als eine komplette Abkehr vom Standard-of-care.
Pirfenidon bleibt eine sinnvolle Alternative, insbesondere bei klassischer IPF und bei Patienten, die Nintedanib nicht tolerieren. Nerandomilast ist wissenschaftlich attraktiv, insbesondere da es sowohl als Monotherapie als auch als Zusatz zu bestehender antifibrotischer Therapie untersucht wurde; für ein deutsches Entwicklungsnarrativ sollte es jedoch bis zur klaren EU-Verfügbarkeit als Zukunftsoption eingeordnet werden.
6. Sicherheits- und Interaktionslogik
Ein Kombinationsansatz aus Nintedanib, Dasatinib/Quercetin und inhalativem Sekretom ist biologisch plausibel, aber sicherheitsseitig anspruchsvoll.
Nintedanib ist mit gastrointestinalen Nebenwirkungen, Leberenzymerhöhungen und Blutungsrisiken assoziiert. Die EMA-Produktinformation weist außerdem auf P-gp-vermittelte Interaktionen hin; starke P-gp-Induktoren können die Nintedanib-Exposition senken, während Inhibitoren die Exposition erhöhen können.
Dasatinib hat ein deutlich anderes Risikoprofil als klassische pneumologische Medikamente. Die EMA-Produktinformation beschreibt die Anwendung als onkologische Therapie; bekannte Risiken umfassen unter anderem Myelosuppression, Infektionen, Blutungen, Pleuraergüsse und pulmonal-arterielle Hypertonie. Das ist für Lungenfibrose besonders relevant, weil Pleuraergüsse, Dyspnoe und pulmonale Gefäßkomplikationen klinisch mit der Grunderkrankung überlappen können.
Quercetin kann CYP3A4- und P-gp-Aktivität beeinflussen; Dasatinib ist CYP3A4- und Transporter-relevant, weshalb pharmakokinetische Interaktionen nicht trivial sind.
Daraus ergibt sich: Die Kombination sollte nicht als dauerhafte Dreifach-Dauertherapie gedacht werden, sondern als kontrolliertes, kurzes, eng überwachtes Senolytika-Fenster innerhalb einer stabilen antifibrotischen Basistherapie. Zu überwachen wären Blutbild, Leberwerte, Gerinnungs-/Blutungsereignisse, EKG/QT-Risiko, Echokardiographie bei Verdacht auf pulmonale Hypertonie, Pleuraergüsse, Infektionen, Sauerstoffbedarf, Exazerbationen und HRCT-Verlauf.
7. Fazit
Lungenfibrose ist mehr als überschießende Kollagenablagerung; sie ist ein chronisch fehlgeleitetes Reparaturprogramm. Ein einzelnes Antifibrotikum kann dieses Programm bremsen, aber selten umkehren. Die Kombination aus moderner antifibrotischer Basistherapie, senolytischem Reset und inhalativem mesenchymalem Stammzellsekretom eröffnet einen rationalen Weg, Progressionskontrolle und Gewebereparatur erstmals systematisch zusammenzuführen. Für Patienten, die unter Lungenfibrose leiden, liegt die besondere Chance darin, daß ein solcher Kombinationsansatz die Behandlung der Lungenfibrose von „Verlangsamung“ in Richtung einer direkten Krankheitsmodifikation verschieben könnte – in der Hoffnung, eine Lungentransplantation zu vermeiden oder zumindest zu verzögern.